Medical Device CE Mark / CE certification Overview
What is CE marking for medical devices?
CE marking on a medical device or invitro diagnostic device shows that the device meets the legal requirements for selling Medical devices in EU market. To get a Medical device CE mark, the manufacturer must document the product’s quality, safety and performance.
Operon Strategist is the best CE Marking Consultant, we provide end to end services to ensure CE marking for medical devices and IVDs.
What is CE Mark Technical File or Design Dossier Compilation and Review?
Compiling your technical file or design dossier is a critical step in Europe’s CE certification process and a requirement for compliance with the Medical Devices Directive 93/42/EEC. In Vitro Diagnostic Medical Devices Directive 98/79/EC, or Active Implantable Medical Devices Directive 90/385/EEC. This has now transitioned to MDR and IVDR.
An incomplete or improperly completed Technical File may result in unexpected delays or even prevent market entry. We Operon Strategist help you in the process of making a defined and comprehensive technical file with all product details required for CE marking. We also provide assistance in your process of making technical files and review it at every step for compliance with CE Mark. We have the technical expertise and experience to provide CE marking services. Our team supports clients in meeting “European submission” standards that declares the product offered is in compliance with the Essential Requirements of relevant European safety, health and Environmental protection regulation.
As CE marking certification consultants we help the new medical device manufacturers to bring forth their medical device in the EU(European market). Our CE experts go in depth of your device design, risk analysis, biological safety, clinical evaluation, testing reports, post market surveillance. CE mark on a product suggests that it mandatorily meets minimum legal requirements of the related directives which allow it to be placed legally on the market in any European member state.
CE is a mark for Health, Safety and Environment protection standards to sell medical products in EEA (European Economic Area), CE mark is recognized worldwide. CE Mark is a declaration by manufacturers that the products they are selling in EEA are meeting the requirements of EC Directives. In order to market products in the European market, the products must have a CE mark which declares that a product meets all relevant European Medical Device Directives.
CE Mark is a declaration by manufacturers that the products they are selling in EEA are meeting the requirements of EC Directives. The Medical Device CE Mark is a conformity mark which all such devices must have before they can be marketed.
- As CE mark consultants for medical devices we’ll help you for the process of making a defined technical file with all the product details.
- We will help you in meeting European submission standards that state the product offered is in compliance with the exact requirements of European safety.
Requirement of CE Marking / Medical Device CE Mark
The era of CE Marking began in 1985. However, it was the year 1993 which embarked the beginning of the Medical Device Directive (93/42/EEC). This was initiated towards an effort to harmonise the regulations pertaining to medical devices with the European Union legislation. CE marking affirms an indication that the manufacturer complies with the relevant EU legislation regardless of the origin of manufacturing. Affixing CE mark to a product declares sole responsibility of the manufacturer to conform to all the regulatory requirements for obtaining a Medical Device CE Mark for the product to permit free trade and sale in the European Economic Area (EEA).
Prior to 1985, many countries within the EEC had different and non-uniform policies and rules in practise which impacted the quality of products in those regions. This resulted in device ambiguity which in turn questioned the safety and efficacy of the devices. A need to regularise and create a homogenous system grew vital to irradiate the technical barriers. This then led to streamlining the consumer protection safety need and thus began the origin of directives and requirement of CE Marking. These directives laid down the Essential Requirements for demonstrating safety and performance uniformly in the EU. Therefore medical device classification for CE marking becomes vital in order to identify requirements.
MDD to New MDR Classification of Medical Devices
The prerequisites for MDR classification for medical devices are basically equivalent to those in the present Medical Devices Directive (MDD). The EU MDR is shaking up the medical device industry and the order rules have not been left immaculate. The MDR decides the congruity assessment course for the device. While the order principally worries the maker, if the device falls into Classes IIa, IIb, or III it has suggestions for the Notified body.
Before medical devices manufacturers can lawfully CE stamp their products in Europe, they should consent to the fitting medical devices order or guideline set out by the EU Commission. It is crucially critical to know the right medical device classification for your product before CE marking your devices. A classification impacts the administrative prerequisites for your devices, just as the endorsement course and its related expenses.
The channel to EU MDR compliance
EU MDR compliance for Medical Device CE Mark.The main basic understanding of the law, the difficulties, with under two years until the check runs out, it might amaze you to discover that 78% of medicinal device organizations don’t yet trust that they have adequate comprehension of the EU MDR enactment.
A large portion of the obstacles organizations are confronting begin here: At an abnormal state, the industry must acknowledge the way that the take-off and translation of any broad-based control will contain components of “hazy area”. The voyage to consistency won’t occur without any forethought and slip-ups will be made by any association endeavouring to agree to the considerable rundown of MDR necessities.
EU IVDR – InVitro Diagnostics Regulation
IVDR Classification – IVD CE mark
The New IVDR Classification necessary changes are compared with the previous In Vitro Diagnostics Directives structure. Before the arrangement of IVDD was a basic and inflexible rundown – based framework that took into account diverse choice by various EU states.
The IVDR (In Vitro Diagnostics Regulation) is the new regulatory reason for placing on the market, making accessible and placing into service in-vitro diagnostics medical devices on the European market. It will supplant the EU’s present order on in-vitro diagnostics medical devices (98/79/EC). As a European guideline, it will be compelling in all EU member states.
IVDR Technical Documentation
The set of IVDR technical documentation is evaluated per device category. IVDR is the new regulatory basis for placing in the market, making it available and putting it into service in-vitro diagnostics medical devices in the European market. In-vitro diagnostics (IVD) is a system where medical devices and reagents are utilized to analyze examples, for example, blood, urine, instruments, tissues, and other body liquids which are derived from the human body to recognize sicknesses, conditions and contaminations.
A portion of the huge innovations consolidated in IVD incorporate polymerase chain response (PCR), microarray procedures, and sequencing innovation, and mass spectrometry, which are utilized for test arrangement. The European Unions In Vitro Diagnostic Regulation (IVDR) 2017/746), which replaces the current In Vitro Diagnostic Directive (IVDD 98/79/EC), presents the risk-based grouping for IVD medical, notwithstanding another necessary technical documentation.
Certification Process
We help you understand and meet the regulation to ensure your product completes the CE Marking process efficiently and successfully. And answer the questions. How to get ce mark for medical device ? and Medical Device CE Mark certification process.
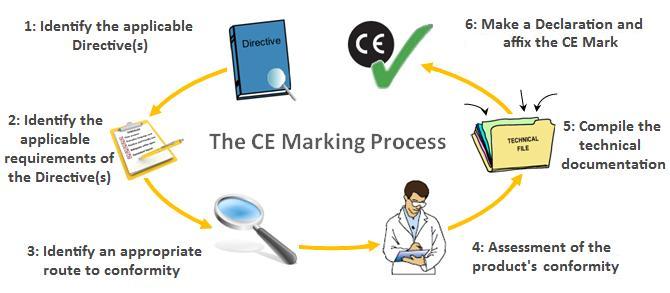
Before the CE Mark may be affixed to a medical device and legally sold within the European Union, the manufacturer or exporter must complete the following:
- Prepare Technical Documentation (Technical File) to show the product’s compliance with applicable essential requirements and conformity assessment procedures of the applicable device directive
- Register their device with the appropriate Competent Authorities
- Receive a device-specific CE Certificate from a Notified Body (Class I devices do not require a certificate from a Notified Body)
- Determine your certification process based on your device class.
- Fulfill the essential requirements – Ensure that your medical device fulfills the essential requirements in Annex I of the Medical Device Directive.
- Establish a monitoring system – As a manufacturer, you are required to monitor your products once they are on the market, in case accidents involving your products occur.
- Establish an accident reporting system – If an accident or near-accident involving any of your products takes place, you are obligated to report this to the authorities.
- Issue a Declaration of Conformity.
- Save the documentation for five years – The new revision of the MDD requires that records for implantable devices be kept for 15 years. Declaration of conformity, technical documentation, reports, and certificates from the Notified Body etc. must be kept for at least five years after the product has been taken out of production.
- Register with the appropriate authorities in Europe.